Modeling of Drugs and Ligands - MoDaL

Modeling of Drugs and Ligands (MoDaL)
Summer School
June 22-26, 2020, Torun, Poland
September 14-18 (the most probably), 2020, Torun, Poland
April or May 2021, Torun, Poland
June 21-25, 2021, Torun, Poland
or
September 2021
According to the difficult coronavirus pandemic situation in Poland we need to postpone MoDaL Summer School. The most probable term of MoDaL school is June or September 2021.
All PhD students who received funding to participate in the MoDaL summer school can take part in it (if they have PhD student status). If someone is unable to attend the school, please contact the PROM coordinator.
Due to the postponement of the BIT20 conference to 2021, the MoDaL summer school will not take place together with this conference. Participants will be provided with other activities related to quantum chemistry and molecular modeling
According to coronavirus issue please do not pay conference fee. We will inform you about ne dates and deadlines soon. We will monitor situation with coronavirus pandemia and will send you information about MoDaL summer school.
Goal: increasing (satisfy) a general interest in the approaches for drug and protein modeling applied in the context of computer-aided drug design and developing the basic skills in the field of computational biochemistry for the unassisted simulation planning and realization in the group of PhD students
Participants of the MoDaL Summer School will take part in practical exercises during the first three and a half days. On Thursday afternoon and Friday they will take part in the BIT20 - BioInformatics in Toruń 2020 conference, which will take place for the twentieth time.
PhD students from outside Poland can apply for nine grants for accommodation and travel costs from the PROM programme iled by the Nicolaus Copernicus University. The amount of co-financing ranges from 5840 PLN (about 1360 EUR) to 9840 PLN (about 2300 EUR) (depending on the distance from Toruń). Applications for co-financing can be submitted on the website of PROM programme http://prom-umk.pl/ from 20 January up to 30 June 2020.
All participants are obliged to show a poster concerning their doctoral dissertation during the BIT20 conference.
In order to ensure the best possible quality of teaching, the number of summer school participants is limited to 23 persons.
Specific objectives:
-
extensive presentation of complementary computational chemistry approaches for computer-aided drug design and ligand design, starting from quantum chemistry in the gas phase via solvent models to ligand docking and molecular dynamics of drug-protein interaction;
-
detailed analysis of structure-property relationship for the selected examples of active molecules and their specific functions in living organisms;
-
introduction to the ways of the in silico modeling of the new drug candidates including the extensive conformational search, electrostatic potentials and presence and steric arrangement of functional groups;
-
demonstration of selected tools for ligand-macromolecule complex modeling and its interaction;
-
discussion of the basic principles of bioactive molecule parametrization;
-
practical realization of the exemplary scientific project devoted to the modeling of selected drugs and ligands, its derivatives and their interaction with a corresponding protein;
Effects:
-
a better understanding of the computational parts of scientific manuscripts devoted to the variety of theoretical approaches;
-
learning of the available techniques and tools for modeling of biomolecules;
-
acquiring skills necessary for an individual planning of computational procedure to be applied for drug/ligand design or protein interaction modeling;
-
gaining the ability of the self-governing;
General outline:
The MoDaL workshop is devoted to the practical application of computational chemistry tools and molecular modeling tools for the practical drug design and description of the ligand-protein interaction.
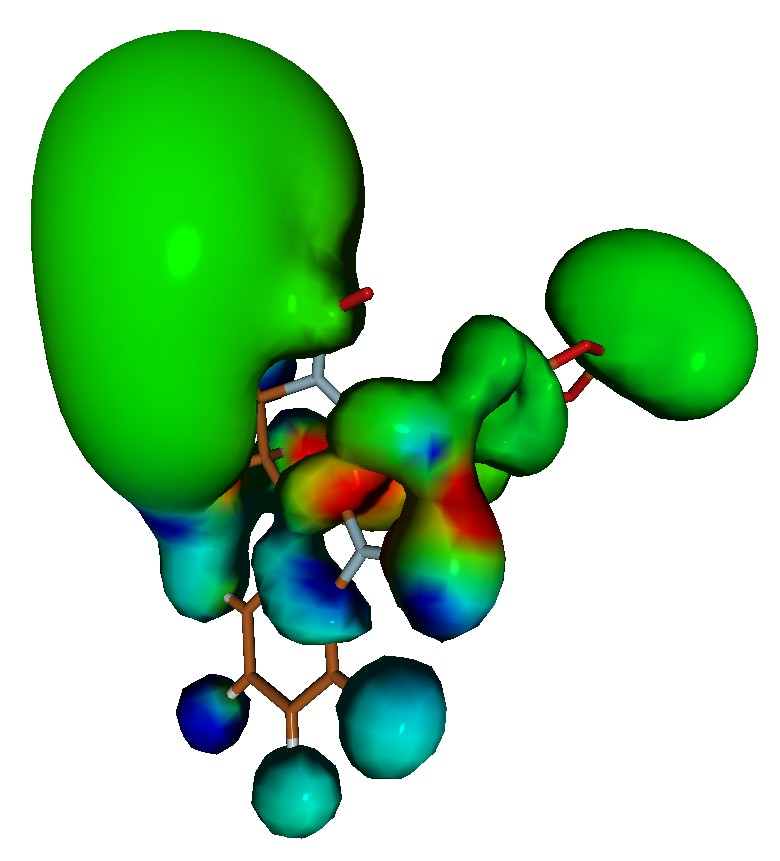
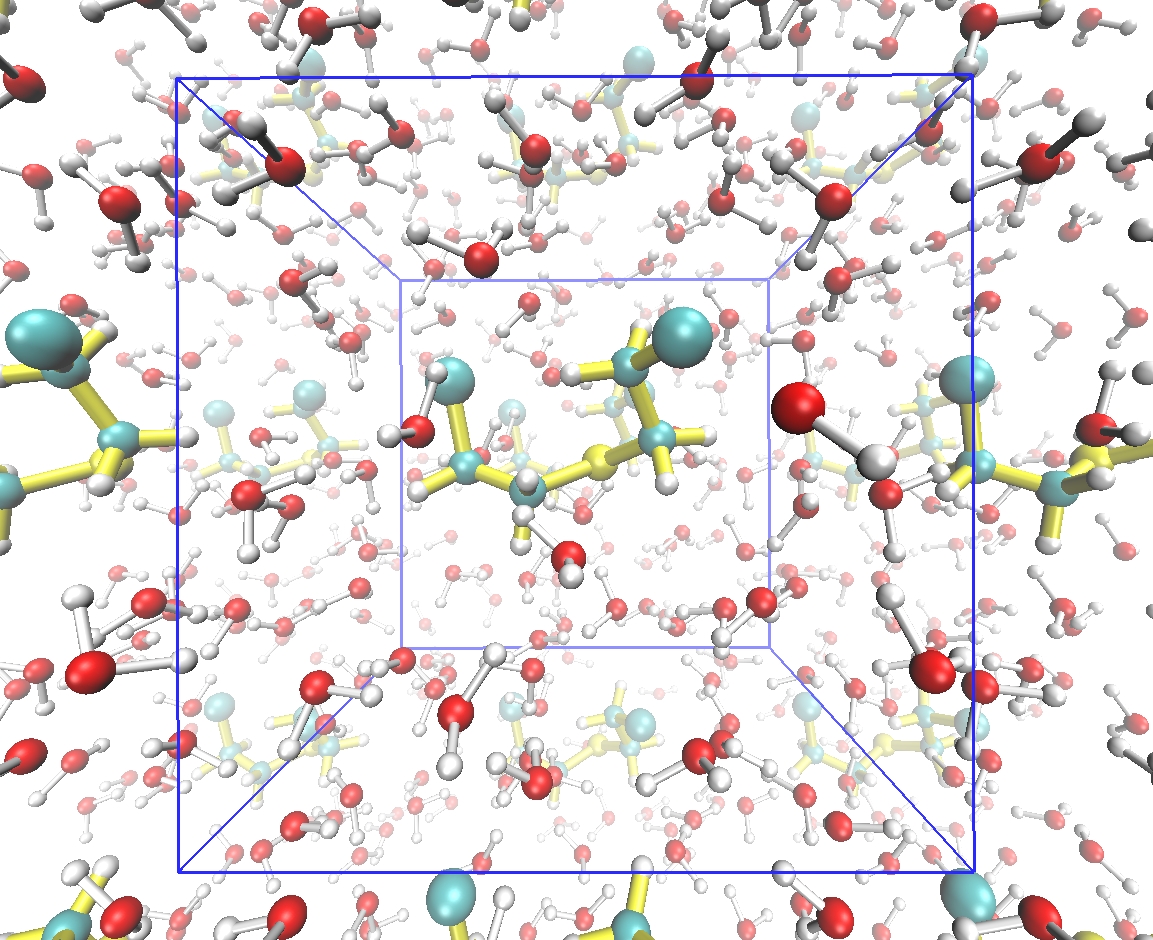
From the molecular point of view, the biological activity of the investigated molecules depends on their characteristic steric and electronic features which allow for their optimal interaction with the receptor. This set of features is commonly known as pharmacophore. Correctly developed pharmacophore model, prepared on the basis of the set of active and inactive molecules, is essential for the drug design based on the ligand structure. However in the actual ligand activity not only its character plays a role, but also its interactions with the environment, namely solvent molecules, ions, other ligands. Thus, the optimal methods and techniques for the theoretical description of intermolecular interactions as well limitations of currently available solvent models and interaction approaches need to be taken into account within the process of the modeling of new drug.
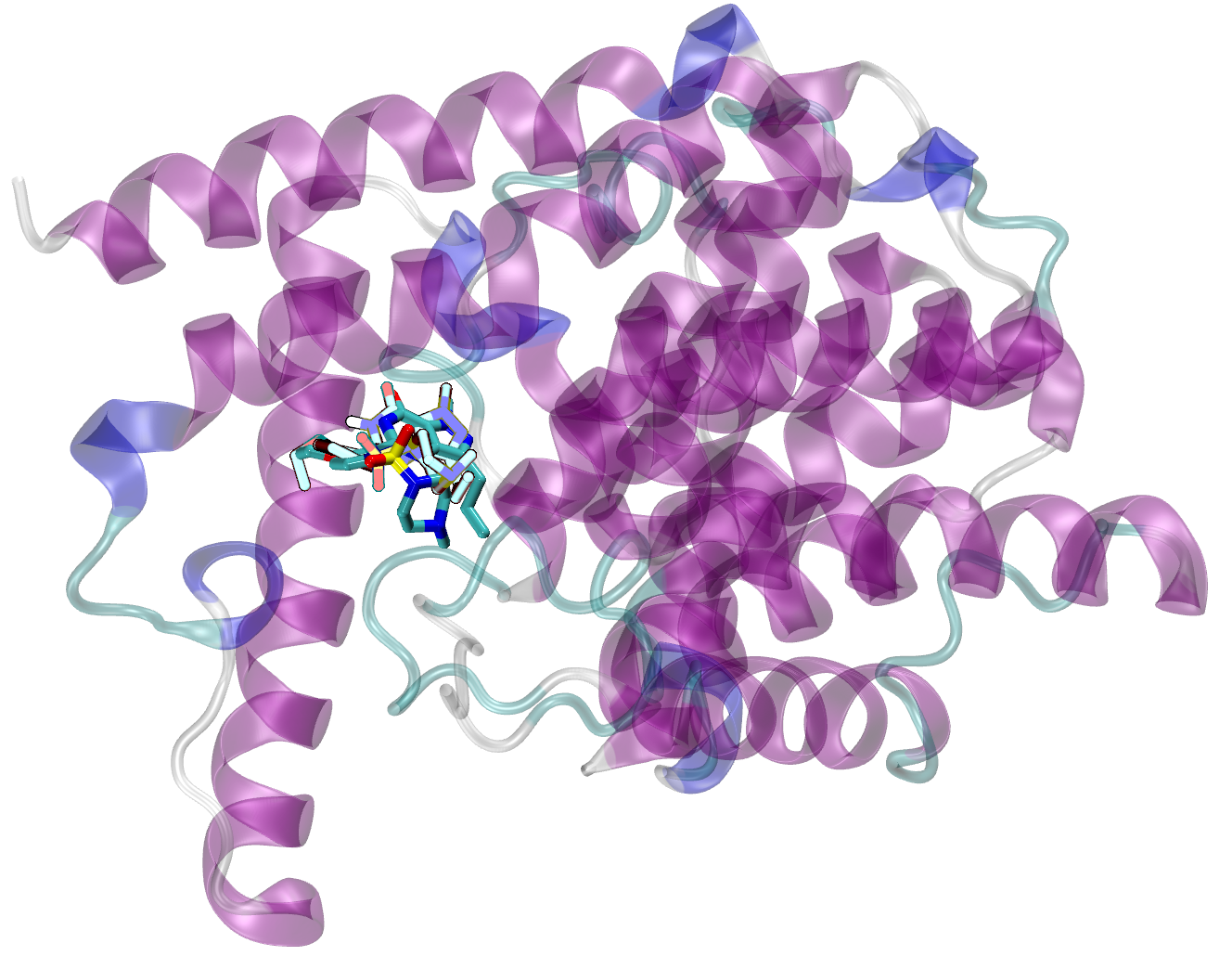
On the other hand, when the three-dimensional receptor structure is known from the X-ray crystallography or NMR spectroscopy, the drug design process focuses on finding the ligands maximizing the mutual interactions with the protein active site. This is the idea of protein-based drug design.
The present workshop will touch upon all of the necessary fields of theoretical calculations, starting from quantum description of an isolated ligand and its interactions with environment via the complex problem of proper solvation treatment, the basic dynamic simulation of a ligand in solvent, docking of the ligand to the active site of a receptor, the small molecules parametrization for the force field application and finally the real molecular dynamics simulations of a ligand-receptor interaction. The easily accessible, preferably free software will be applied in order to enable for a simple organization of own infrastructure for further calculations after the workshop.
 |
 |
 |
 |
http://www.ccl.net/chemistry/a/conferences/
Dates: June 21, 2021 - May 24, 2020
Location: Toruń
Conference menu
- Main page
- Important Dates
- Fees
- Registration
- Program
- Accommodation
- Venue/Travel
- Organization & contact
Organizers:
![]()


![]()
![]()
Sponsors:
